
戈谢病(Gaucher disease, GD)是一种由葡萄糖脑苷脂酶(GBA)基因变异引起的常染色体隐性遗传性溶酶体贮积病。2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,戈谢病被收录其中。
11月11日,晋城市人民医院举办了 “戈谢病多学科诊疗中心” 的正式成立大会,此诊疗中心设立的目的在于推动医院对戈谢病诊治的规范化进程。




什么是戈谢病?
戈谢病(Gaucher disease, GD)是一种由葡萄糖脑苷脂酶(GBA)基因变异引起的常染色体隐性遗传性溶酶体贮积病,表现为多系统的脂质沉积, 受累的血液、肝脾、骨骼及神经系统功能受损而出现相应的临床表现,导致肝脾肿大、贫血、血小板减少、骨痛以及骨骼病变,发育迟缓而可能终身残疾甚至死亡。

戈谢病发病机制图解
戈谢病的患病率是多少?
在全球范围内,戈谢病的新生儿标化发病率为(0.39~5.80)/10万,患病率为(0.70~1.75)/10万。东欧和中欧犹太人的患病率为118/10万。中国尚无大样本量流行病学统计数据,华东(上海)地区和台湾地区开展的戈谢病新生儿筛查研究显示发病率分别约为1/80 855和1/10 313。
戈谢病有哪些类型和临床表现?
根据神经系统是否受累及进展速度,分为非神经病变型(Ⅰ型)、急性神经病变型(Ⅱ型)、慢性或亚急性神经病变型(Ⅲ型)3种亚型。

儿童戈谢病的临床分型(表格源自网络)
如何识别戈谢病?
戈谢病表现为多脏器功能受累,最常见最明显的症状为肝脾肿大,临床表现为“大肚子”,可有血小板减少、贫血等症状,部分有骨痛,生长迟缓的表现。II型/III型戈谢病患儿还会出现神经病变,表现为认知障碍、精神运动发育落后、癫痫等。
因为戈谢病有多种表现,家长发现患儿有肚子大、骨痛、发育落后、贫血等表现时要及时就诊,需要及时至医院就诊,尽早诊断及治疗。

儿童戈谢病主要特征图解
如何诊断戈谢病?
儿童戈谢病的检查方法包括酶活性检测、基因检测、骨髓形态学检查、生物标志物检测、影像学检查及其他辅助检查方法。其中,酶活性检测是戈谢病诊断的金标准。除酶活性检测外,也建议患儿及亲属及时完善GBA基因检测,可以检测致病基因携带情况,明确变异类型,并进行家系验证,有助于遗传咨询和产前诊断。
戈谢病的主要治疗方法
戈谢病目前主要的治疗方法有特异性和非特异性治疗。特异性治疗包括酶替代治疗、造血干细胞移植、底物抑制疗法以及基因治疗。
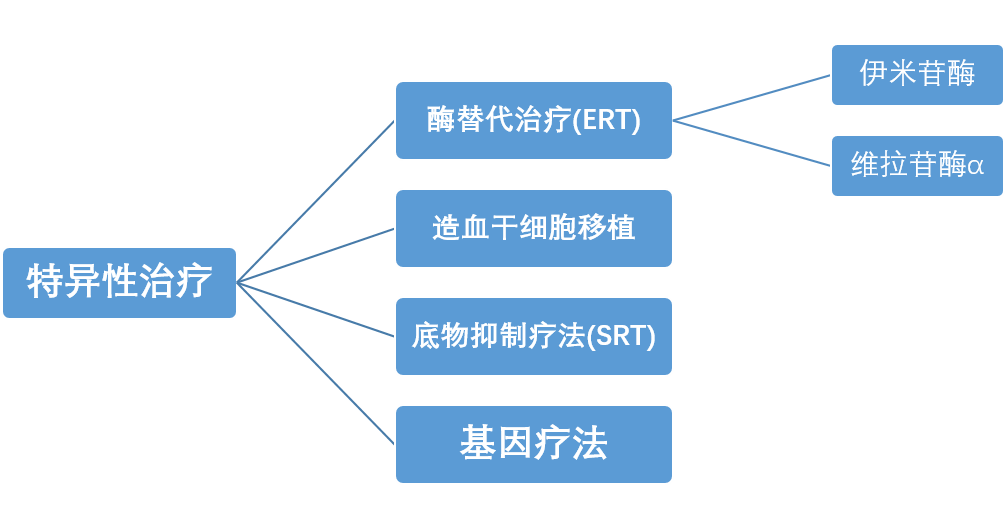
戈谢病的特异性治疗

戈谢病的非特异性治疗
戈谢病的基因疗法
基因治疗是输注以病毒为载体的基因治疗产品,给药后可特异性转导靶器官,并在靶器官中长期表达GCase蛋白,GCase蛋白可分泌至血液中并被其他脏器如脾脏、肺脏、骨髓等器官吸收。戈谢病患者体内各脏器蓄积的有害糖脂代谢物会被GCase蛋白降解,从而有效治疗戈谢病。

基因治疗的基本原理
如何面对戈谢病?
戈谢病是一种常染色体隐性遗传病,如果头胎生育了戈谢病患者,那么夫妻再次生育,其子女患病的风险是25%。因此,对有曾生育过戈谢病患者的家庭,应在再次怀孕前进行遗传咨询,必要时可以通过第三代试管婴儿(PGT-M),或者产前诊断基因检测,从源头上阻断戈谢病的发生,打破家族遗传病的“魔咒”。
免责声明:以上图文部分来源于网络,如有侵权请与我们联系,将及时删除。
版权所有:晋城市人民医院
地址:晋城市城区白水东街1666号
24小时咨询热线:0356-96559
jcsrmyyyb@126.com
048026
ICP证:晋ICP备12005875号-1 晋卫网审[2014]第0007号
技术支持:晋城市云祥大数据科技运营有限公司

扫一扫关注我们